The effect of nucleic acid modifications on digestion by DNA exonucleases
by Greg Lohman, Ph.D., New England Biolabs, Inc.
New England Biolabs offers a wide variety of exonucleases with a range of nucleotide structure specificity. Exonucleases can be active on ssDNA and/or dsDNA, initiate from the 5´ end and/or the 3´ end of polynucleotides, and can also act on RNA. Exonucleases have many applications in molecular biology, including removal of PCR primers, cleanup of plasmid DNA and production of ssDNA from dsDNA. In this article, we explore the acitivity of commercially available exonucleases on oligonucleotides that have chemical modifications added during phosphoramidite synthesis, including phosphorothioate diester bonds, 2´-modified riboses, modified bases, and 5´ and 3´ end modifications. We discuss how modifications can be used to selectively protect some polynucleotides from digestion in vitro, and which modifications will be cleaved like natural DNA. This information can be helpful for designing primers that are stable to exonucleases, protecting specific strands of DNA, and preparing oligonucleotides with modifications that will be resistant to rapid cleavage by common exonuclease activities.
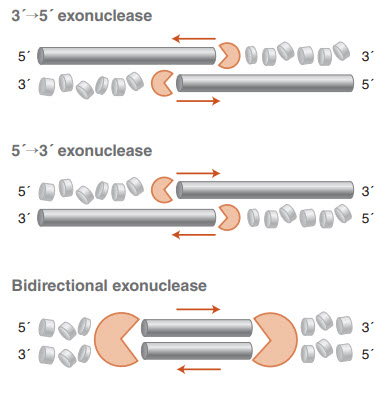
The ability of nucleases to hydrolyze phosphodiester bonds in nucleic acids is among the earliest nucleic acid enzyme activities to be characterized (1-6). Endonucleases cleave internal phosphodiester bonds, while exonucleases, the focus of this article, must begin at the 5´ or 3´ end of a nucleic acid strand and cleave the bonds sequentially (Figure 1). Exonucleases may be DNA or RNA specific, and can act on single-stranded or double-stranded nucleic acids, or both. Double-strand specific exonucleases may initiate at blunt ends, nicks, or short single-stranded 5´ or 3´ overhangs, though most exonucleases are active on a subset of these structures. For a summary of the substrate specificity of exonucleases available from NEB, view our selection chart, Properties of Exonucleases and Non-specific Endonucleases.
A variety of DNA exonucleases have been characterized from many different organisms; in vivo, these enzymes play critical roles in polynucleotide repair, recycling, error correction, and protection from exogenous DNA (6-8). In vitro, exonucleases are used in many applications where it is desirable to remove certain nucleic acids. For example, Exonuclease V (RecBCD) (Exo V, NEB #M0345) is often used to remove contaminating linear ssDNA and dsDNA from plasmid preparations (4,9); T7 Exonuclease (T7 Exo, NEB #M0263) can be used to generate 3´ overhangs in DNA (4, 10, 11); Exonuclease I (Exo I, NEB #M0293), Thermolabile Exonuclease I (NEB #M0568) or Exonuclease VII (Exo VII, NEB #M0379) can be used to eliminate ssDNA PCR primers, leaving double-stranded products undigested (12, 13), and Lambda Exonuclease (Lambda Exo, NEB #M0262) can be used to convert dsDNA to ssDNA for a variety of applications (14-16). More information on common applications of exonucleases available from NEB can be found in our selection chart, Common Applications of Exonucleases and Non-Specific Endonucleases.
What about cases where you only want to degrade some of the ssDNA in a reaction? Or, when you want to make ssDNA from a dsDNA substrate, but which strand is degraded matters greatly? What about cases where the ends of your nucleic acids are modified—will exonucleases still digest the substrate, or cleave the modification? Several methods depend on selective protection of polynucleotides, such as protection of primers from degradation by polymerase exonuclease domains (17), selective protection of one strand of a DNA duplex for the production of ssDNA (14-16), and the protection of polynucleotides from degradation by serum nucleases, as in the case of RNA interference drugs (18, 19). In each of these cases, it is critical to understand the influence of modifications on exonuclease activity—which modifications inhibit nucleotide cleavage and which do not.
Recently, researchers at NEB have worked to characterize the interaction between exonucleases and modified polynucleotides, as part of a broader effort to gain deeper insight into the sequence and structural determinants of nuclease activity and specificity. In an effort to catalog the modifications that inhibit exonuclease digestion, we have treated polynucleotides containing a range of modifications (including non-standard bases, sugars and backbone chemistries) with exonucleases under the recommended in vitro reaction conditions. This article will summarize data from the literature, as well as the key results from NEB’s work related to understanding the activity of exonucleases on chemically modified polynucleotides. We will focus on the most widely used—and most successful—method for blocking nuclease activity, the phosphorothioate bond (20-23), but will also discuss the use of other modifications to inhibit nuclease activity, as well as which modifications have little to no effect on exonuclease digestion.
Phosphorothioate linkages
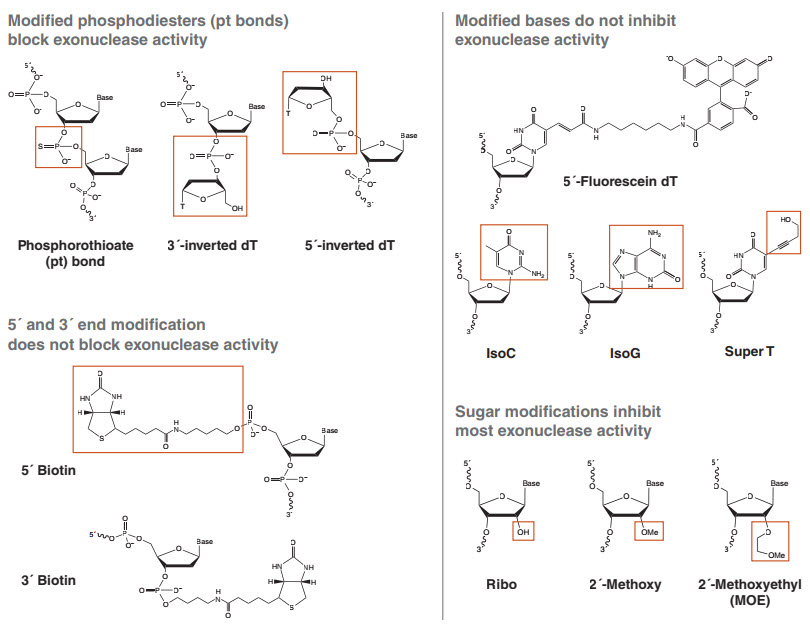
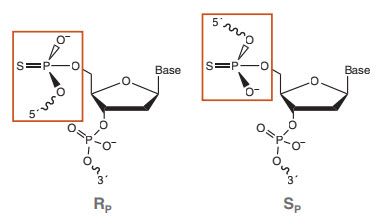
A phosphorothioate (pt) bond is a phosphodiester linkage where one of the two non-bridging oxygens has been replaced by a sulfur (Figure 2). This modification has been used for decades to inhibit nuclease phosphodiesterase and phosphoryl transferase activities, as well as for gaining mechanistic insights into these enzymes (20, 23). Chemically, the substitution of oxygen with sulfur does not dramatically change the reactivity of the bond, and pt-containing polynucleotides can still function in many enzymatic reactions. In a typical phosphodiester bond, the two non-bridging oxygens are chemically equivalent. When one of these oxygens is replaced by sulfur, however, the phosphorus is now connected to four distinct groups, rendering it a chiral center with two possible configurations referred to as “SP” and “RP” (Figure 3). It is this key feature that confers resistance for the majority of nuclease enzymes studied; one configuration will react at rates similar to a phosphodiester, while the other is significantly inhibitory or completely unreactive. Isomer reactivity varies from enzyme to enzyme, and different pt isomers can inhibit enzymes that catalyze the same reaction (e.g., phosphoryl transfer). For example, DNA Polymerase I (DNA Pol I, NEB #M0209) can incorporate deoxynucleotide triphosphates with a pt ester at the α phosphate (dNTPαS), allowing formation of pt-bonded polynucleotides. However, it can only react with SP configured dNTPαS molecules, and does so with inversion of the stereocenter to form exclusively RP-configured pt bonds in the product. Conversely, the 3´→ 5´exo activity of this polymerase cleaves RP but not SP configured bonds (20). Alternatively, the 3´→ 5´ exo activity of E. coli Exonuclease III (Exo III, NEB #M0206) cleaves SP but not RP configured pt bonds (24). Therefore, DNA created from the incorporation of dNTPαS by DNA Pol I is highly resistant to exonuclease cleavage by Exo III (25).
Phosphorothioates can block many, but not all, exonucleases. To block exonuclease cleavage, the pt bonds must be placed at the end(s) where the enzyme initiates, e.g., the 5´ end for Lambda Exo and the 3´ end for Exo III. It is important to note that a single pt bond is insufficient to fully protect an oligonucleotide from exonuclease digestion. When the pt bond is installed via an oxidation step during phosphoramidite synthesis, a nearly equal amount of each isomer (SP and RP) is formed at each pt linkage (20). Since most enzymes can cleave one of these isomers, a single chemically installed pt will protect only half the molecules from digestion by a given exonuclease. Thus, it is typically recommended that 3–6 pt bonds be used to block exonuclease digestion, to prevent this read-through. One might expect that because each bond is a 50:50 mixture of isomers, when presented with 5 consecutive isomers, a given enzyme could cleave the first bond on half the molecules, then half of the molecules that had the first bond hydrolyzed would have the second hydrolyzed, and so on, such that there would be a range of partially degraded products. In practice, it has been reported (and confirmed by recent results at NEB) that five consecutive pt bonds completely block all exonuclease activity at all pt bond positions (16). The exact reasons for this are not currently known, but it is likely that exonucleases engage multiple bases at once, and the net effect of the isomeric mixture somehow prevents the active site from properly organizing around bonds that are the normally cleavable pt isomer.
There are several commonly used exonucleases that are not blocked even by 5 consecutive pt bonds; for example, Exo V, Exo VII and T5 Exonuclease (T5 Exo, NEB #M0663) all can cleave, leaving short oligos instead of cutting at every bond in a series, and thus can digest DNA by skipping over termini blocked by multiple pt bonds and cleaving at the first phosphodiester (5, 26). Importantly, any enzyme with endonuclease activity, like DNase I, will simply ignore the ends and degrade the polynucleotides from the inside out (unless every phosphodiester bond is replaced by a phosphorothioate). Keeping these important exceptions in mind, phosphorothioate bonds remain the most generally applicable (and relatively inexpensive) way to protect oligonucleotides from digestion by exonucleases. For a complete list of DNA exonucleases and their interaction with pt bonds, view our selection chart, Properties of Exonucleases and Non-specific Endonucleases.
2´-modified nucleosides
Generally, DNA exonucleases do not digest RNA portions of oligonucleotides, though RNA is itself susceptible to RNases and nonspecific hydrolysis. We have further found that hybridizing RNA to DNA strands does not block the activity of dsDNA exonucleases on the DNA strand. Hybridization of ssDNA to RNA will block the activity of ssDNA exonucleases as effectively as hybridization to dsDNA. Additionally, certain 2´-O-modified riboses, are both stable to spontaneous hydrolysis and offer strong resistance to exonuclease activity (27). 2´-O-methyl and 2´-O-methoxyethyl (MOE) nucleosides, which contain bulky substituents off the sugar ring, have been shown to grant strong resistance to nucleases and additionally increase the strength of annealing to complementary DNA and RNA. These features have found utility in antisense nuclease strategies, to make oligonucleotides that are both resistant to degradation and able to bind
tightly to target RNAs.
These sugar modifications also work in vitro to block exonuclease activity quite strongly. Our studies have found that, while a single terminal MOE nucleoside only weakly inhibits exonuclease activity, three successive MOE modifications provide enhanced resistance to many exonucleases, including Exo I, Exo III, Lambda Exo, RecJF (NEB #M0264) and polymerase exonucleases activities, such as that of DNA Polymerase I, Large (Klenow) Fragment (NEB #M0210). Similar to pt bonds, several exonucleases can digest through these regions, notably T5 Exo, T7 Exo, Exo V, Exo VII and Exo VIII. Overall, exonuclease inhibition by MOE is quite strong, but pt bonds are more effective and are cheaper to install for most manufacturers. However, if for some reason the pt chemistry is not desired, 2´-O-modified ribose moieties are a viable alternative.
Other 5´/3´ end modifications
Several other modifications, such as the inverted deoxythymidine bases and dideoxynucleotides (Figure 2) have been reported to suppress serum nuclease activity when appended to the end of synthetic oligonucleotides (27). Many other modifications may be attached through “linkers” at either the 5´ or 3´ end, including fluorescent tags, biotin or other affinity labels, or reactive groups for attachment to beads or surfaces. These linkers are typically connected to the 5´ or 3´ end via a phosphodiester. What is the interaction of these modified ends with exonucleases?
We have surveyed a range of these modifications under typical in vitro exonuclease assays. In general, while many provide modest inhibition as compared to a 5´-phosphate, all exonucleases tested could cleave all modifications connected through phosphodiester bonds. Interestingly, this poor inhibition held true for the inverted dT modifications, which have been reported to grant extra stability versus degradation by serum exonucleases for aptamers and other modified oligonucleotides. In our hands, 3´-inverted dT blocked only the relatively weak 3´→ 5´ exonuclease activity of DNA Polymerase I, Large (Klenow) Fragment (NEB #M0210) and Exonuclease T (Exo T, NEB #M0265), but did not block more active exonucleases such as in T7 DNA Polymerase (NEB #M0274), Exo I or Exo III. Similarly, 5´-inverted dT partially inhibited only Lambda Exo activity, which is known to require a 5´-phosphate for efficient initiation. Other 5´→ 3´ exonucleases were not significantly inhibited by this modification, showing complete digestion after a one-hour incubation under the recommended usage conditions.
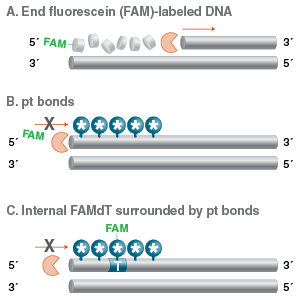
We do not recommend 5´/3´ end modification as a good strategy for producing nucleotides resistant to exonuclease degradation in vitro. Researchers should be aware that these modifications will be cleaved by the majority of exonucleases, potentially leading to the loss of fluorescent labels and affinity tags. If a modification stable to exonuclease activity is needed, a better strategy is to use internal labels connected to the 5-methyl position of dT (e.g., Fluorescein dT, Figure 2). If these modified dT bases are used near the end of an oligo, they can be protected with surrounding pt bonds (Figure 4). The linkage to the base is not susceptible to enzymatic cleavage, and the pt bonds will protect the backbone from digestion, as described above.
Base modifications
None of the exonucleases available from NEB were significantly inhibited by modified bases under the conditions we tested. Modifications tested included 5-methyl-substituted dT (e.g., Fluorescein dT), deoxyuridine, the Tm-enhancing “super T,” and the non-natural base pair isoG:isoC (Figure 2) (28). All modified substrates were digested completely by all the exonucleases tested. Some modifications showed weak blockage, pausing at the modification site before completely degrading the substrate. For several exonucleases tested, modified dT bases with large modifications off the 5-methyl position (Figure 2) showed a buildup of partially-digested intermediates, apparently stalling just before the modification; in no case did this resistance approach the inhibition seen for 2´ MOE sugars or pt linkages.
Conclusion
We have evaluated a variety of chemical modifications for their inhibition of exonuclease activity at the 5´ and 3´ ends of oligonucleotides. Broadly, the phosphorothioate modification, one of the more well-known used modifications to block nuclease cleavage, remains the most effective choice to protect oligonucleotides from degradation. However, one must be careful to use multiple pt bonds, place them at the correct end of the oligonucleotide to match the polarity of the exonucleases used, and be aware that several exonucleases can read-through or bypass terminal pt bonds; your choice of nucleases is as important as the modifications used. Aside from pt bonds, MOE nucleotides are the next best choice for providing nuclease resistance in vitro, with similar caveats to pt bonds. The vast majority of end modifications, including affinity tags and fluorophores, as well as internal non-standard bases, provide little if any nuclease resistance, and will be cleaved completely in vitro.
View a PDF of this feature article
References
- Lehman, I. R., and Nussbaum, A. L. (1964) J. Biol. Chem. 239, 2628-2636.
- Nichols, N. M. (2011) Curr. Protoc. Mol. Biol. Chapter 3, Unit3.12.
- Richardson, C. C., Lehman, I. R., and Kornberg, A. (1964) J. Biol. Chem. 239, 251-258.
- McReynolds, L. A., and Nichols, N. M. (2011) Curr. Protoc. Mol. Biol. Chapter 3, Unit 3.11.
- Lovett, S. T. (2011) EcoSal Plus 4.
- Yang, W. (2011) Q. Rev. Biophys. 44, 1-93.
- Bebenek, A., and Ziuzia-Graczyk, I. (2018) Curr. Genet. 64, 985-996.
- Tsutakawa, S. E., Lafrance-Vanasse, J., and Tainer, J. A. (2014) DNA Repair (Amst). 19, 95-107.
- Karu, A. E., MacKay, V., Goldmark, P. J., and Linn, S. (1973) J. Biol. Chem. 248, 4874-4884.
- Kerr, C., and Sadowski, P. D. (1972) J. Biol. Chem. 247, 305-310.
- Straus, N. A., and Zagursky, R. J. (1991) Biotechniques. 10, 376-384.
- Li, H. H., Cui, X. F., and Arnheim, N. (1991) Nucleic. Acids. Res. 19, 3139-3141.
- Enzymatic PCR Cleanup using Exonuclease I and Shrimp Alkaline Phosphatase. New England Biolabs, Ipswich, MA.
- Civit, L., Fragoso, A., and O’Sullivan, C. K. (2012) Anal. Biochem. 431, 132-138.
- Murgha, Y. E., Rouillard, J. M., and Gulari, E. (2014) PLoS One 9, e94752.
- Nikiforov, T. T., Rendle, R. B., Kotewicz, M. L., and Rogers, Y. H. (1994) PCR Methods Appl. 3, 285-291.
- Skerra, A. (1992) Nucleic Acids Res. 20, 3551-3554.
- Evers, M. M., Toonen, L. J., and van Roon-Mom, W. M. (2015) Adv. Drug Deliv. Rev. 87, 90-103.
- Lundin, K. E., Gissberg, O., and Smith, C. I. (2015) Hum. Gene Ther. 26, 475-485.
- Eckstein, F. (1985) Annu. Rev. Biochem. 54, 367-402.
- Spitzer, S., and Eckstein, F. (1988) Nucleic Acids Res. 16, 11691-11704.
- Eckstein, F., and Gish, G. (1989) Trends Biochem Sci 14, 97-100.
- Eckstein, F. (2014) Nucleic Acid Ther. 24, 374-387.
- Putney, S. D., Benkovic, S. J., and Schimmel, P. R. (1981) Proc. Natl. Acad. Sci. U S A, 78, 7350-7354.
- Yang, Z., Sismour, A. M., and Benner, S. A. (2007) Nucleic Acids Res. 35, 3118-3127.
- Sayers, J. R., and Eckstein, F. (1990) J. Biol. Chem. 265, 18311-18317.
- Kratschmer, C., and Levy, M. (2017) Nucleic Acid Ther. 27, 335-344.
- Piccirilli, J. A., Krauch, T., Moroney, S. E., and Benner, S. A. (1990) Nature, 343, 33-37.

