Polymerase Fidelity: What is it, and what does it mean for your PCR?
The discovery and development of high-fidelity polymerases has for many years been a key focus at New England Biolabs (NEB). Highfidelity amplification is essential for experiments whose outcome depends upon the correct DNA sequence (e.g., cloning, SNP analysis, NGS applications). Whereas traditional fidelity assays are sufficient for Taq and other moderately faithful enzymes, Q5, an ultra highfidelity enzyme, pushes the limits of current methods used to assess this critical feature of DNA polymerases.
Krist N. Hausken, Ph.D., John A. Pezza, Ph.D., Rebecca Kucera, M.S., Luo Sun, Ph.D., New England Biolabs, Inc.
What does DNA polymerase fidelity mean?
Maintaining sequence integrity of a newly copied DNA strand relative to its template during DNA replication is critical for the accurate transfer of genetic material from one generation of cells to the next. The fidelity of DNA replication, which is the accuracy with which the DNA sequence is copied, is maintained by the action of DNA polymerases, enzymes responsible for adding nucleotides to the new DNA strand. Fidelity comparisons between polymerases can be expressed in absolute terms, often by the number of errors per 1,000 or 10,000 nucleotides, or relative terms by using Taq DNA polymerase as the reference standard (1X).
Mechanisms of polymerase fidelity
Accurate DNA replication involves multiple steps, including the ability to read the template base, select the appropriate nucleoside triphosphate and insert the correct nucleotide such that Watson-Crick base pairing is maintained. However, replicative DNA polymerases make mistakes and can incorporate the incorrect nucleotide, resulting in mutations relative to the template. To prevent errors, DNA polymerases have evolved mechanisms that allow them to detect and correct mistakes before they become permanent mutations in the DNA. The geometry of the polymerase active site determines selection of the correct incoming nucleotide and aligns the catalytic groups to ensure efficient incorporation. If an incorrect nucleotide does bind in the active site, incorporation is slowed due to the sub-optimal architecture of the active site complex. This lag time increases the opportunity for the incorrect nucleotide to dissociate before polymerase progression, thereby allowing the process to start again, with a correct nucleoside triphosphate (1,2).
Some polymerases have a 3´→5´ exonuclease (AKA proofreading) domain that confers additional protection against incorporation of the wrong nucleotide. Proofreading involves the enzymatic removal of incorrect nucleotides from the 3’ end of the growing DNA strand before they become permanently incorporated. The perturbation and lag time caused by mispaired bases is detected and the polymerase moves the 3´ end of the growing DNA chain into the proofreading 3´→5´ exonuclease domain. There, the incorrect nucleotide is removed by the 3´→5´ exonuclease activity, whereupon the chain is moved back into the polymerase active site for addition of the correct nucleotide.
Measurement of polymerase replication fidelity
Polymerase fidelity assays take many forms and have been used extensively for comparing high-fidelity polymerases. The pioneering work of Thomas Kunkel (3) utilized portions of the lacZα gene in M13 bacteriophage to correlate host bacterial colony color changes with errors in DNA synthesis. While these assays were high-throughput, they depended on phenotypic selection and therefore could not resolve single-base errors.
Similar to the Kunkel assays, Wayne Barnes (5), utilized 16 cycles of PCR to copy the entire lacZ gene and portions of two drug resistance genes with subsequent ligation, cloning, transformation and blue/white colony color determination. Most errors in the lacZ-encoding β-galactosidase gene cause a loss-of-function ability to utilize the Xgal substrate on agar plates, resulting in a white colony phenotype that indirectly measured fidelity even after correction for error propagation during PCR. The entire 1.9 kb gene was amplified but only 349 bases produce a color change (6), obscuring accurate detection of polymerase error rate. As a more direct read-out of fidelity, Sanger sequencing of individual, cloned PCR products offered the advantage that all mutations could be detected (7), and as the cost for sequencing dropped over time the number of targets and reads increased the accuracy of error detection.
Previously at NEB, a modification of the Barnes assay (5) utilizing a 1000 amino acid open reading frame was used to determine mutation rates using both the blue/white selection method after 16 PCR cycles (Figure 1A) and by Sanger sequencing after 25 PCR cycles (Figure 1B), and the error rate per base incorporated was determined after calculating the effective number of amplification cycles for each experiment (5,8). Comparing the data sets from Taq indicated that the two methods generated similar results, with error rates of ~1 in 3,500 nucleotides from 215,000 nucleotides sequenced. Q5 High-Fidelity DNA Polymerase, on the other hand, yielded a significantly lower number of errors than Taq in both assay systems; only two errors of the 440,000 nucleotides sequenced were identified, consistent with an error rate of around 1 in 1,000,000 nucleotides. Significantly more sequencing reads would be necessary to accurately quantify the error rate of proofreading polymerases, but this would also decrease the throughput of the workflow.
Next-generation sequencing platforms overcame this hurdle by providing vast sequencing data on the order of millions to billions of read nucleotides, allowing measurement of a statistically significant number of polymerase errors. However, the lower threshold for determining polymerase error rates by barcoded Illumina sequencing was reported as 1 × 10−6 errors/base, which is still within range of the error rate for high fidelity polymerases (9, 10, 11, 12). We have utilized a PacBio single-molecule (SMRT) sequencing assay to accurately and directly sequence PCR products to capture the various types of errors generated during PCR (13). With SMRT sequencing, PCR products can be directly sequenced without molecular indexing or an intermediary amplification step (as is needed for Illumina sequencing), and accuracy is achieved by sequencing the same molecule multiple times and deriving a highly accurate consensus sequence for each read that can be used to identify true replication error rates. By sequencing plasmid DNA, which is virtually devoid of nucleotide errors, we found the background error rate for the SMRT sequencing fidelity assay to be 9.6 × 10−8 errors/base, making it an appropriate method to quantify the fidelity of proofreading polymerases. Additionally, because this assay measured fidelity in a cell-free system (no transformation), all errors associated with template replication could be identified, including the number of errors (substitutions, indels) per base per doubling event, template switching and cruciform structures intrinsic to the amplicon, PCR-mediated sequence recombination, and non-enzymatic DNA damage induced during thermal cycling.
Results
To validate the use of single-molecule sequencing for measuring replication fidelity of a LacZ amplicon, we measured the fidelity of Taq DNA polymerase using both PacBio SMRT- and traditional Sanger sequencing (Figure 1C). The errors/base/doubling were similar for SMRT (1.8 x 10-4) and Sanger (1.3 x 10-4) sequencing, despite SMRT producing over two orders of magnitude more reads (3.58 × 107 nucleotides) than Sanger (3.23 × 105 nucleotides).
SMRT-seq was able to generate enough sequencing reads to determine polymerase fidelity for several common high-fidelity DNA polymerases with statistical accuracy (Figure 1 C). The base substitution rates spanned 3 orders of magnitude with the highest fidelity observed for Q5 High-Fidelity DNA Polymerase (5.3 × 10−7 sub/base/doubling; 280X Taq) and the lowest for exonuclease-deficient Deep Vent (exo-) polymerase (5.0 × 10−4 sub/base/doubling; 0.3X Taq). The contribution of proofreading activity to fidelity was determined by comparing exonuclease-proficient Deep Vent DNA Polymerase to the exonuclease-deficient Deep Vent (exo-), where we found that the presence of the 3´-5 exonuclease domain provided a 125-fold decrease in error rate from 5.0 × 10−4 to 4.0 × 10−6 sub/base/doubling.
Fidelity measurements by different methods
A. Blue/White Colony Screening
| DNA Polymerase | Substitution rate | Accuracyb | Fidelity, rel. to Taqc | White/Total colonies |
| Taq | ~2.7 x 10-4 | 3,700 | 1 | 17,589/30,192 |
| Q5 | ~1.4 x 10-6 | 710,000 | 193 (±101) | 119/22,296 |
B. Sanger Sequencing
| DNA Polymerase | Substitution ratea | Accuracyb | Fidelity, rel. to Taqc | Total bases |
| Taq | ~3 x 10-4 | 3,300 | 1 | ~215,000 |
| Q5 | ~1 x 10-6 | 1,000,000 | ~300 | ~440,000 |
C. PacBio SMRT Sequencing
| DNA Polymerase | Substitution ratea | Accuracyb | Fidelity, rel. to Taqc | Total bases |
| Taq | 1.5 × 10-4 (± 0.2 × 10-4) | 6,456 | 1 | 98,396,789 |
| Q5 | 5.3 × 10-7 (± 0.9 × 10-7) | 1,870,763 | 280 | 112,619,228 |
| Phusion | 3.9 × 10-6 (± 0.7 × 10-6) | 255,118 | 39 | 118,262,939 |
| Deep Vent | 4.0 × 10-6 (± 2.0 × 10-6) | 251,129 | 44 | 106,217,940 |
| Pfu | 5.1 × 10-6 (± 1.1 × 10-6) | 195,275 | 30 | 79,614,976 |
| PrimeSTAR GXL | 8.4 × 10-6 (± 1.1 × 10-6) | 118,467 | 18 | 118,964,566 |
| KOD | 1.2 × 10-5 (± 0.2 × 10-5) | 82,303 | 12 | 121,234,438 |
| Kapa HiFi HotStart ReadyMix | 1.6 × 10-5 (± 0.3 × 10-5) | 63,323 | 9.4 | 101,742,963 |
| Deep Vent (exo-) | 5.0 × 10-4 (± 0.1 × 10-4) | 2,020 | 0.3 | 60,218,605 |
a.Reported error rates are per base per doubling as detailed in Materials and Methods. Standard deviations were determined based on sequencing several samples and are given here in brackets.
b.Accuracy is calculated as 1 over substitution rate such that accuracy is a number of bases over which 1 substitution error is expected.
c.Fidelity relative to Taq numbers are computed separately for each amplicon (LacZ-1, LacZ-2, DNA-1, DNA-2) and the average number is reported per DNA polymerase.
Q5® DNA Polymerase possesses higher fidelity than other polymerases assayed by SMRT sequencing
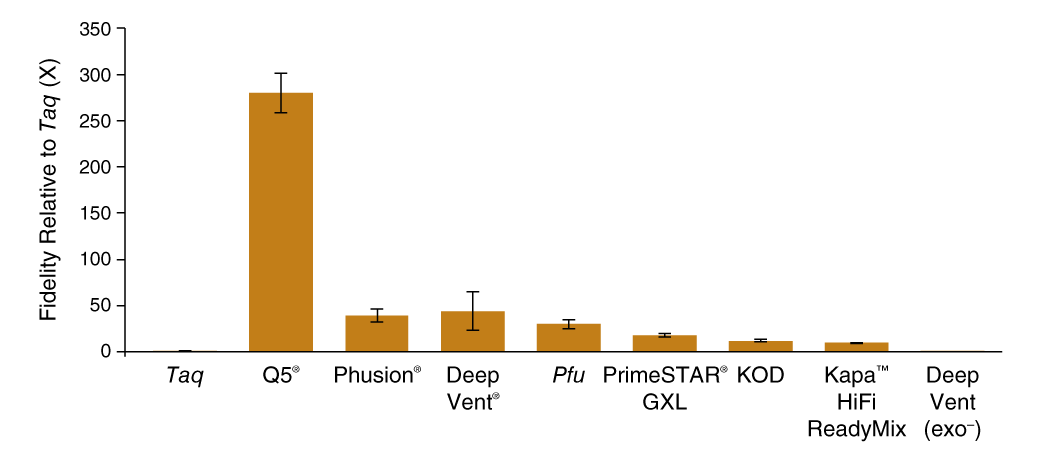
Comparison of base substitution error rates of various DNA polymerases relative to Taq DNA polymerase generated using the PacBio SMRT Seq assay.
Conclusion
In conclusion, a single-molecule sequencing assay was used to measure the frequency of various types of PCR errors, from single-nucleotide base substitutions to large-scale template switching events. Single-molecule sequencing has lowered the threshold for error rate determination, yet as engineered DNA polymerases achieve higher accuracy, other mutagenic sources start to become significant contributors to mutations in PCR products. For example, in the case of Q5 High-Fidelity DNA Polymerase, DNA damage induced by thermocycling was two-fold higher than polymerase misincorporation. The amount of mutagenic DNA damage introduced during thermocycling, identified primarily as deoxyuridine resulting from cytosine deamination, was measured to be 1.4 × 10−6 per base per PCR cycle, which can be a significant contribution to the total errors generated during amplification.
As PCR errors are propagated during exponential amplification, seemingly infrequent events can have profound implications for downstream analysis, particularly for next-generation sequencing. Understanding the role that persistent DNA damage plays in contributing to mutations in PCR products will help to identify and avoid false sequence information, and single-molecule sequencing can yield further insight into the polymerase behavior responsible for such errors.
References
- Johnson, K.A. (2010) Biochemica et Biophysica Acta, 1804, 1041–1048.
- Joyce, C.M. and Benkovic, S.J. (2004) Biochemistry, 43, 14317–14324.
- Kunkel, T.A. and Tindall, K.R. (1988) Biochemistry, 27, 6008–6013.
- Ling, L.L. et al. (1991) Genome Research, 1, 63–69.
- Barnes, W.M. (1992) Gene, 112, 29–35.
- Provost, G.S. et al. (1993) Mutation Research-Fundamental and Molecular Mechanisms of Mutagenesis, 288, 133-149.
- McInerny, P. et al. (2014) Molecular Biology International, 8, 1-8.
- Eckert, K.A. and Kunkel, T.A. (1991) PCR Methods and Applications, 1, 17–24.
- Schmitt, M.W. et al. (2012) Proc. Natl. Acad. Sci, 109(36), 14508-14513.
- Kinde, I. et al. (2011) Proc. Natl. Acad. Sci, 108(23), 9530-9535
- Hestand, M.S. et al. (2016) Mutat. Res., 784-785, 49-45.
- Lee, D.F. et al. (2016) Nucleic Acids Res., 44(13), e118.
- Potapov, V. and Ong, J.L. (2017) PLoS ONE, 12(1), e0169774.


