Protocol for High Molecular Weight DNA (HMW DNA) Extraction from Bacteria (NEB #T3060)
Below is a detailed protocol for the extraction of high molecular weight DNA from bacteria samples. Please refer to the complete product manual for more detailed guidance. For help selecting input amounts, visit Choosing Input Amounts for the Monarch HMW DNA Extraction Kits. If you are using this kit upstream of ultra-long DNA sequencing on the Oxford Nanopores Technologies platform, please also review our Ultra-Long DNA Sequencing Workflow®’ Guidance for HMW DNA Extraction Upstream of Oxford Nanopore Technologies as well as our Protocol for UHMW DNA Cleanup in the Oxford Nanopore Technologies® UL Library Prep Workflow.
Materials Required but Not Supplied:
- Microcentrifuge
- Thermal mixer containing a 1.5 ml tube block (optional: 2 ml tube block for elution)
- Recommended: vertical rotating mixer (e.g., Thermo Scientific® HulaMixer® Sample Mixer).
- Ethanol (≥ 95%).
- Cold PBS, 300 µl per sample (Low Input: 150 µl per sample). Alternatively, TE or Tris buffer can be used.
- Isopropanol, 550 µl per sample (Low Input: 275 µl/sample).
- 1.5 ml DNase-free, low DNA binding microfuge tubes (e.g., Eppendorf® DNA LoBind®, #0030108051) are recommended for elution and storage (1 per sample); it is especially important to use low DNA binding tubes if working with UHMW DNA, which tends to bind to plastic surfaces.
- For Gram-negative bacteria: Lysozyme (25 mg/ml, 10 µl per sample)
- For Gram-positive bacteria: STET Buffer (Current Protocols in Molecular Biology) containing Lysozyme (10 mg/ml) can be an effective lysis agent (150 µl or 300 µl per sample).
- Additional lysis agents may be required (e.g., lysostaphin).
Important Notes Before You Begin:
- Review the complete protocol before beginning.
- Preheat thermal mixer with 1.5 ml block to 37°C, (if available, preheat another to 56°C).
- Add ethanol (≥ 95%) to the gDNA Wash Buffer as indicated on the bottle label.
- Rotor-stator homogenization may be used to obtain shorter gDNA fragments (50–250 kb), which often results in better ligation-based nanopore sequencing results.
- Proteinase K and RNase A should be stored at -20°C.
Starting Material Notes:
The volumes indicated in the protocol vary depending on input amounts. Refer to the following designations throughout the protocol to determine the appropriate volumes
PROTOCOL DESIGNATION |
NUMBER OF BACTERIAL CELLS* |
|
Standard Input |
E. coli: 1 x 109 – 5 x 109 |
|
Low Input |
E. coli: 5 x 108 – < 1 x 109 |
* Optimal input amounts for other bacteria may vary depending on the strain, genome size, and growth conditions.
– E. coli typically produces HMW gDNA in the range of 50 kb – ≥ 500 kb
– B. cereus typically produces HMW gDNA in the range of 50 kb – ≥ 350 kb
HMW gDNA Purification Consists of Two Stages:
PART 2: HMW gDNA Binding and Elution
PART 1: Bacterial Lysis
- Pellet bacterial cells in a Monarch Pestle Tube by centrifugation at maximum speed (> 12,000 x g) for 1 minute. If using a rotor-stator homogenizer, use a compatible 2 ml tube, not included.
- Gram-negative and Gram-positive bacteria are processed differently for the initial lysis steps. The use of bead beating is not recommended as it will result in a significant reduction of gDNA size.
Gram-negative Bacteria
- Resuspend pellet in 300 µl (Low Input: 150 µl) cold PBS. Cold TE or Tris buffer may be used in place of
PBS if preferred.
- Add 10 µl Lysozyme (25 mg/ml, not provided) and mix by vortexing briefly.
- Add 300 µl (Low Input: 150 µl) HMW gDNA Tissue Lysis Buffer to the sample and mix by inverting 5-10 times.
- Incubate at 37°C in a thermal mixer with agitation at the desired speed. The speed of the thermal mixer influences fragment length and lysis time. For most applications, maximum agitation speed (1400–2000 rpm) is recommended. For maximum gDNA size, agitate at 500 rpm. Incubation is complete when lysate turns clear, which is approximately 3–5 minutes for E. coli. At 500 rpm, lysis may take longer.
Gram-positive Bacteria
- Resuspend pellet in 300 µl (Low Input: 150 µl) of an appropriate lysis buffer containing a lytic enzyme and mix by
vortexing briefly. STET buffer with freshly added lysozyme (10 mg/ml) works well for some Bacillus species.
- Incubate at 37°C for 30 minutes (no agitation).
- Add 300 µl (Low Input: 150 µl) HMW gDNA Tissue Lysis Buffer to the sample and mix by inverting 5–10 times.
- Resuspend pellet in 300 µl (Low Input: 150 µl) cold PBS. Cold TE or Tris buffer may be used in place of
PBS if preferred.
- If working with a single thermal mixer, increase the temperature to 56°C. Following lysozyme treatment at 37°C, increase the temperature of the block in the thermal mixer to 56°C.
- Add 20 µl (Low Input: 10 µl) of Proteinase K and mix by inverting 10–20 times.
- Homogenization can be carried out using one of two methods, depending on the desired gDNA size: in a thermal mixer or with a rotor-stator homogenizer. If using a rotor-stator homogenizer, the sample must be in a 2 ml tube (not provided).
Thermal Mixer (for gDNA 50 kb up to ≥ 500 kb)
-
Incubate at 56°C for 30 minutes in a thermal mixer at the desired speed. The speed of the thermal mixer influences fragment length and lysis; higher agitation speeds reduce DNA size and sample lysis time. For most applications, including the standard ligation-based Oxford Nanopore Technologies (ONT) sequencing protocols, maximum agitation speed (1400–2000 rpm) is recommended to produce DNA fragments predominantly 50–250 kb.
To achieve maximum gDNA size, up to the Mb range, use a low agitation speed. Agitation at speeds < 500 rpm is not recommended as gDNA will be significantly tangled, which reduces the efficiency of protein removal in later steps. This tangled DNA is also difficult to dissolve during lysis and elution and can result in visible DNA aggregates.
Rotor-stator Homogenizer (for gDNA 50–250 kb)
-
Within a 2 ml tube, insert the tip of the homogenizer probe and turn on to the lowest setting. Homogenize 5–15 seconds; stop when foam begins to form in the lysate. Additional homogenization may be required to reach optimal gDNA size. gDNA size can be verified by pulsed field gel electrophoresis or FEMTO Pulse. Rotor-stator homogenizers may run at higher speeds after extended use; reduce homogenization time if necessary.
- Transfer to a 1.5 ml Pestle Tube. Incubate at 56°C for a minimum of 30 minutes in a thermal mixer at maximum speed (1400–2000 rpm).
-
- Add 10 µl (Low Input: 5 µl) of RNase A and mix by inverting 5–10 times. Incubate for 10 minutes at 56°C with agitation in a thermal mixer at the speed used in Step 5.
- Change the heat block in the thermal mixer to accommodate a 2 ml tube, and preheat the block to 56°C. If a 2 ml tube block is not available, continue working with the 1.5 ml block.
- Add 300 µl (Low Input: 150 µl) of Protein Separation Solution. Mix by inverting for 1 minute. Alternatively, a vertical rotating mixer at 20 rpm can be used.
- Centrifuge for 10 minutes at 16,000 x g. If working with multiple samples, during centrifugation, prepare the plastics for Part 2, as indicated in the following step. The sample will separate into a large, clear upper phase (DNA) and a lower, clear phase (protein, usually on the bottom of the tube, but occasionally floating). There may also be a white precipitate at the bottom of the tube. Additional centrifugation time (10-20 minutes) may be required for complete phase separation, particularly when low agitation speeds were used.
- If working with multiple samples, prepare and label the plastics for the upcoming steps. Each sample will require:
- Monarch Collection Tube II (no need to label)
- 1 Monarch Bead Retainer inserted into the collection tube; this will be used to remove the wash buffer from the gDNA bound to the beads.
- 2 Monarch 2 ml Tubes; one for phase separation and one for elution.
- 1 1.5 ml microfuge tube (DNA low bind recommended, not provided); this will be used to collect the eluate.
- Using a 1000 µl (Low Input: 200 µl) wide-bore pipette tip, transfer the upper phase containing the DNA (large, clear phase) to a labeled Monarch 2 ml Tube. A substantial fraction of HMW DNA will be located at the interface between the clear upper phase and the protein phase; highest yields will be achieved by transferring as much of the upper phase as possible. Using a 200 µl wide-bore pipette tip to transfer the final volume of the upper phase is recommended for maximum yield. Avoid transferring material from the protein layer, though a small amount (1–2 µl) will not be detrimental. If protein enters the pipette tip, gently push it back into the tube. If a lower protein phase is not visible, leave ~30 µl behind to ensure protein is not carried over. Typically, the transferred volume will be ~ 800 µl (Low Input: ~400 µl). If the volume of the sample is < 700 µl (Low Input: < 350 µl), adjust the volume of isopropanol used in Step 2 of Part 2: HMW gDNA Binding and Elution to 0.7 volumes.
PART 2: HMW gDNA Binding and Elution
- Using clean forceps, add 2 DNA Capture Beads to each sample, which should be contained in a Monarch 2 ml Tube.
- Add 550 µl (Low Input: 275 µl) isopropanol, close the cap, and mix on a vertical rotating mixer at 10 rpm for 5 minutes to attach DNA to the beads. If a vertical rotating mixer is not available, invert slowly and gently by hand 25–30 times. A manual inversion is complete when the tube returns to the upright position. Slow inversion is critical for the DNA to bind to the beads; each full inversion should take ~5–6 seconds. If necessary, flick the tube to release any beads that stick to the bottom of the tube.
After a 2–3 inversions, the solution becomes more viscous and the DNA will wrap loosely around the beads. During the following inversions, precipitation of gDNA may be visible, especially with larger input samples. The DNA complex will often contain small air bubbles. With increasing number of inversions, the DNA will completely wrap around the beads, often causing the beads to stick together. DNA binding to the beads should be complete after 25–30 inversions, and the solution should no longer be viscous. Additional inversions may be necessary for larger input samples.
- Remove and discard liquid by pipetting. Avoid removing any of the gDNA wrapped around the glass beads. For optimal DNA solubility, avoid letting the bound DNA dry out on the beads during this and the following steps; add the next buffer quickly. There are two suggested options for carrying out this step:
- Keeping tube upright, insert pipette tip and gently push beads aside to remove liquid.
- Angle the tube so that beads remain at the bottom, and liquid reaches toward tube opening. Pipette from the liquid surface and continue to angle as liquid is removed (tube will be almost horizontal at the end).
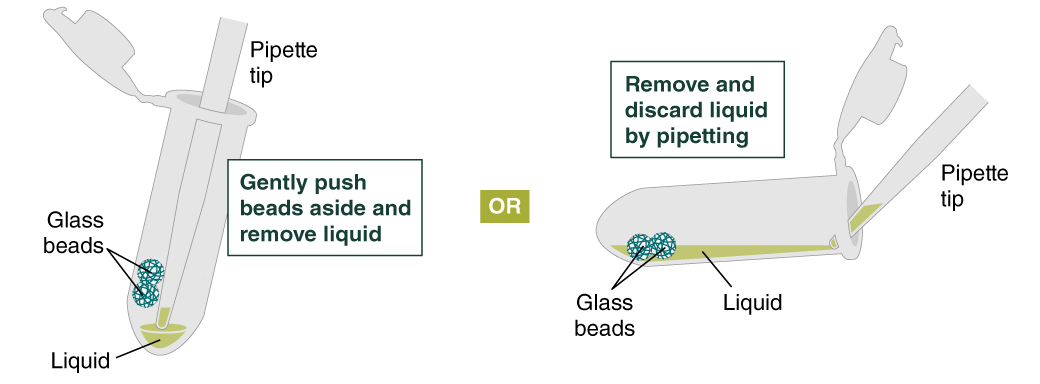
- Keeping tube upright, insert pipette tip and gently push beads aside to remove liquid.
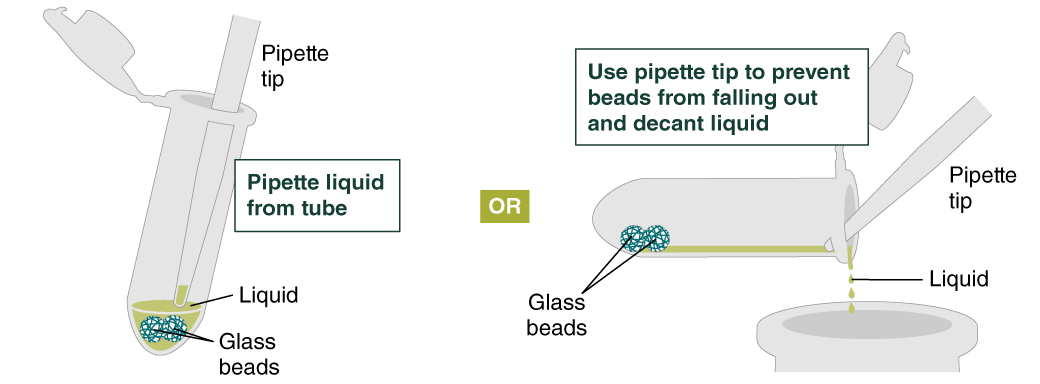
- Add 500 µl gDNA Wash Buffer, close the cap, and mix by inverting the tube 2–3 times. Remove the gDNA Wash Buffer as described in step 3. The loose gDNA complex will condense around the beads more tightly.
- Repeat the wash in Step 4 and remove the gDNA Wash Buffer by pipetting. Alternatively, the buffer can be removed by decanting: position a pipette tip at the top of the angled tube to prevent the beads from falling out. It is not necessary to remove all the gDNA Wash Buffer at this point.
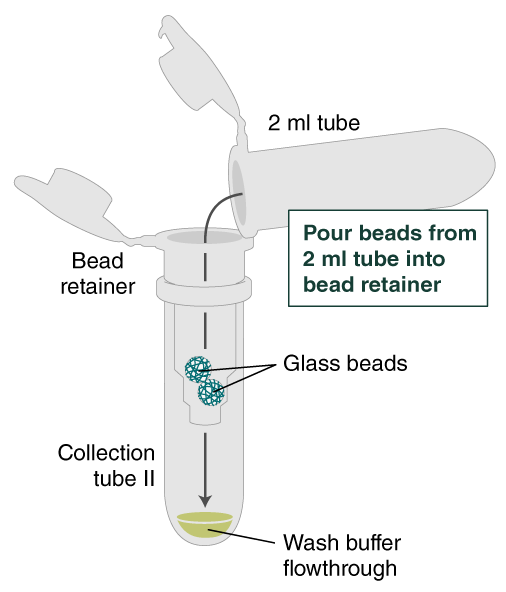
- Place a labeled bead retainer into a Monarch Collection Tube II. Pour the beads into the bead retainer and close the cap. Discard the used Monarch 2 ml Tube. When working with multiple samples, be sure to close the cap of the bead retainer after each transfer of beads.
- Pulse spin (≤ 1 second) the sample in a benchtop minicentrifuge to remove any residual wash buffer from the beads.
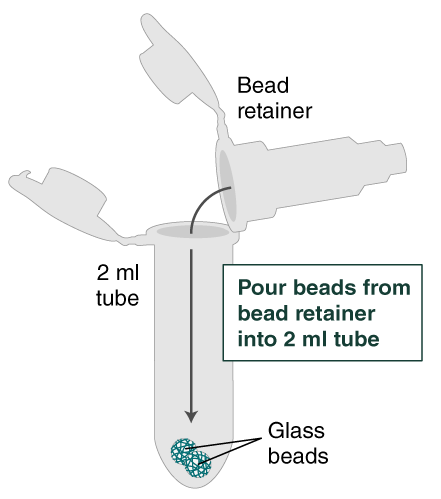
- Separate the bead retainer from the collection tube, pour the beads into a new, labeled Monarch 2 ml Tube, and insert the used bead retainer into the labeled 1.5 ml microfuge tube (DNA low bind recommended, not provided) for later use during elution. Discard the used collection tube.
- Immediately add 100 µl Elution Buffer II onto the glass beads and incubate for a minimum of 5 minutes at 56°C in a thermal mixer with agitation at the lowest speed (300 rpm). Halfway through the incubation, ensure the beads are not stuck to the bottom of the tube by tilting the tube almost horizontally and gently shaking. This ensures that the beads can move freely, allowing for optimal release of the DNA from the beads. It also ensures that the lower bead does not stick to the bottom of the tube during the following transfer step. Elution volume can be reduced to as low as 50 μl without affecting recovery. However, if using < 100 μl, the gentle shaking of the sample should be done several times during the incubation to ensure complete wetting of the beads.
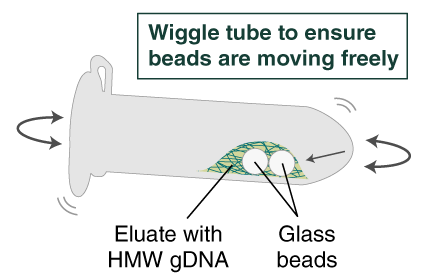
- Ensure the bead retainer is inserted into the 1.5 ml microfuge tube. Pour the eluate and the glass beads into the bead retainer and close the cap. When working with more than 1 sample, it is important to close the cap after each transfer of beads. Typically, all the eluate flows into the bead retainer upon pouring. If any volume remains in the 2 ml tube, spin briefly and transfer.
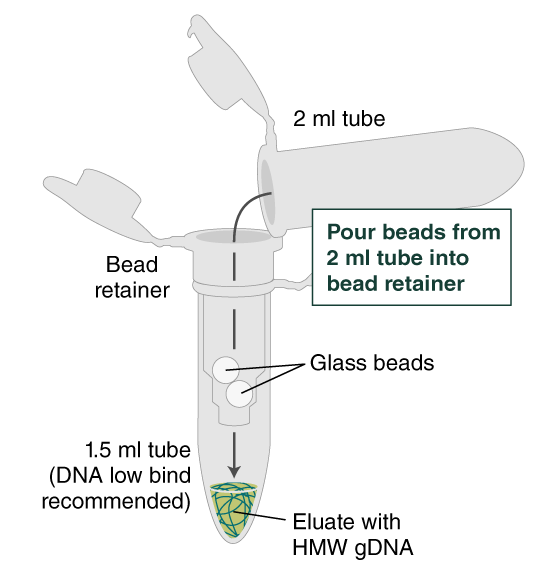
- Centrifuge for 30 seconds at 12,000 x g to separate the eluate from the glass beads. Discard the beads and retainer.
- Pipette eluate up and down 5–10 times with a wide bore pipette tip and ensure any visible DNA aggregates are dispersed. Before analysis or downstream use, HMW DNA must be homogeneously dissolved. After pipetting, incubate at 37°C for 30-60 minutes, overnight at room temperature, or for > 24 hours at 4°C. Pipette up and down 5-10 times again before analyzing or using the HMW DNA. Samples processed using low agitation speeds during lysis will require additional time to fully dissolve. See additional guidance in “Homogenization of HMW DNA Samples”. Samples can be stored at 4°C for short term use (weeks), or at -20°C for long term storage. The elution buffer (10 mM Tris, pH 9.0, 0.5 mM EDTA) is formulated for long term storage of gDNA.

