Protocol for use with Purified mRNA or Ribosome Depleted RNA - NEBNext Ultra Directional RNA Library Prep Kit for Illumina (E7420)
Symbols
 |
This is a point where you can safely stop the protocol and store the samples prior to proceeding to the next step in the protocol. |
 |
This caution sign signifies a step in the protocol that has two paths leading to the same end point but is dependent on a user variable, like the type of RNA input. |
 |
Colored bullets indicate the cap color of the reagent to be added |
This protocol has been optimized using Universal Human Reference Total RNA.
3.1 RNA Fragmentation, Priming and First Strand cDNA Synthesis
RNA fragmentation is only required for intact or partially degraded RNA. Recommended fragmentation times can be found in Table 3.1. Follow protocol in 3.1A to set up the reaction. For highly degraded RNA (FFPE Samples) which do not require fragmentation proceed to Step 3.1B.
3.1A RNA Fragmentation and Priming Starting from Intact or Partially Degraded RNA:
3.1A.1. Set up the following reaction and mix by gentle pipetting:
Purified mRNA or Ribosome depleted RNA (10-100 ng) 5 μl
 (pink) NEBNext First Strand Synthesis Reaction Buffer 4 μl
(pink) NEBNext First Strand Synthesis Reaction Buffer 4 μl
(pink) Random Primers 1 μl
-----------------------------------------------------------------------------------------------------
Final Volume 10 μl
3.1A.2. Incubate the sample at 94°C following the recommendations in Table 3.1 for fragments sizes ~200 nt.
Table 3.1. Suggested fragmentation times based on RIN number of RNA input.
| RNA TYPE | RIN | FRAG. TIME |
| Intact RNA | >7 | 15 min. at 94°C |
| Partially Degraded RNA | 2–6 | 7–8 min. at 94°C |
3.1A.3. Transfer the tube to ice.
First Strand cDNA Synthesis
Dilute Actinomycin D stock solution (5 μg/μl) to 0.1 μg/μl in nuclease free water for immediate use.
Note: Dilute solutions of Actinomycin D are very sensitive to light. In solution, Actinomycin D tends to absorb to plastic and glass. For these reasons, unused dilute solutions should be discarded and not stored for further use. However, frozen aliquots of a concentrated stock solution (5 μg/μl) in DMSO are expected to be stable for at least a month at -20°C.
(pink) Murine RNase Inhibitor 0.5 μl
Actinomycin D (0.1 μg/μl) 5 μl
(pink) ProtoScript II Reverse Transcriptase 1 μl
Nuclease free water 3.5 μl
----------------------------------------------------------------------
Final volume 20 μl
Note: If you are following recommendations in Appendix A, for longer RNA fragments, increase the incubation at 42°C from 15 minutes to 50 minutes in Step 5.
3.1A.5. Incubate the sample in a preheated thermal cycler (with the heated
lid set at 105°C) as follows:
10 minutes at 25°C
15 minutes at 42°C
15 minutes at 70°C
Hold at 4°C
3.1A.6. Proceed directly to Second Strand cDNA Synthesis, Step 3.2.1.
3.1B Priming of Highly Degraded RNA (FFPE) which has a RIN ≤ 2 and does not Require Fragmentation:
3.1B.1. Set up the following priming reaction and mix by gentle pipetting:
Purified mRNA or Ribosome depleted RNA (10-100 ng) 5 μl
(pink) Random Primers 1 μl
-----------------------------------------------------------------------------------------------------
Final volume 6 μl
3.1B.2. Incubate the sample in a preheated thermal cycler as follows:
5 minutes at 65°C, with heated lid set at 105°C. Hold at 4°C.
3.1B.3. Transfer the tube directly to ice.
First Strand cDNA Synthesis
Dilute Actinomycin D stock solution (5 μg/μl) to 0.1 μg/μl in nuclease free water for immediate use.
Note: Dilute solutions of Actinomycin D are very sensitive to light. In solution, Actinomycin D tends to absorb to plastic and glass. For these reasons, unused dilute solutions should be discarded and not stored for further use. However, frozen aliquots of a concentrated stock solution (5 μg/μl) in DMSO are expected to be stable for at least a month at -20°C.
3.1B.4. To the primed RNA from Step 3.1B.3 (6 μl) add the following components and mix by gentle pipetting:
(pink) NEBNext First Strand Synthesis Reaction Buffer 4 μl
(pink) Murine RNase Inhibitor 0.5 μl
Actinomycin D (0.1 μg/μl) 5 μl
(pink) ProtoScript II Reverse Transcriptase 1 μl
Nuclease free water 3.5 μl
-----------------------------------------------------------------------------------------------------
Final volume 20 μl
Note: If you are following recommendations in Appendix A, for longer RNA fragments, increase the incubation at 42°C from 15 minutes to 50 minutes in Step 5.
3.1B.5. Incubate the sample in a preheated thermal cycler (with the heated
lid set at 105°C) as follows:
10 minutes at 25°C
15 minutes at 42°C
15 minutes at 70°C
Hold at 4°C
3.1B.6. Proceed directly to Second Strand cDNA Synthesis, Step 3.2.1
3.2 Perform Second Strand cDNA Synthesis
3.2.1. Add the following reagents to the First Strand Synthesis reaction (20 μl):
Nuclease-free water 48 μl
 (orange) Second Strand Synthesis Reaction Buffer 8 μl
(orange) Second Strand Synthesis Reaction Buffer 8 μl
(orange) Second Strand Synthesis Enzyme Mix 4 μl
-----------------------------------------------------------------------------------------------------
Total volume 80 μl
3.2.2. Mix thoroughly by gentle pipetting.
3.2.3. Incubate in a thermal cycler for 1 hour at 16°C, with heated lid set at ≤ 40°C.
3.3 Purify the Double-stranded cDNA Using 1.8X Agencourt AMPure XP Beads
3.3.1. Vortex AMPure XP Beads to resuspend.
3.3.2. Add 144 μl (1.8X) of resuspended AMPure XP Beads to the second strand synthesis reaction (~80 μl). Mix well on a vortex mixer or by pipetting up and down at least 10 times.
3.3.3. Incubate for 5 minutes at room temperature.
3.3.4. Quickly spin the tube in a microcentrifuge to collect any sample on the sides of the tube. Place the tube on an appropriate magnetic rack to separate beads from supernatant. After the solution is clear (about 5 minutes), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain DNA targets.
3.3.5. Add 200 μl of freshly prepared 80% ethanol to the tube while in the magnetic rack. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
3.3.6. Repeat Step 3.3.5 once for a total of 2 washing steps.
3.3.7. Air dry the beads for 5 minutes while the tube is on the magnetic rack with lid open.
Caution: Do not over dry the beads. This may result in lower recovery of DNA target.
3.3.8. Remove the tube from the magnet. Elute the DNA target from the beads into 60 μl 0.1X TE Buffer or 10 mM Tris-HCl. Mix well on a vortex mixer or by pipetting up and down. Quickly spin the tube and incubate for 2 minutes at room temperature. Place the tube in the magnetic rack until the solution is clear.
3.3.9. Remove 55.5 μl of the supernatant and transfer to a clean nuclease free PCR tube.
Note: If you need to stop at this point in the protocol samples can be stored at –20°C.
3.4 Perform End Prep of cDNA Library
3.4.1. Mix the following components in a sterile nuclease free tube:
Purified double-stranded cDNA (Step 3.3.9) 55.5 μl
(green) NEBNext End Repair Reaction Buffer (10X) 6.5 μl
(green) NEBNext End Prep Enzyme Mix 3 μl
-----------------------------------------------------------------------------------------------------
Total volume 65 μl
3.4.2. Incubate the sample in a thermal cycler (with the heated lid set at 75°C) as follows:
30 minutes at 20°C
30 minutes at 65°C
Hold at 4°C
3.4.3. Proceed immediately to Adaptor Ligation.
3.5 Perform Adaptor Ligation
Dilute the  (red) NEBNext Adaptor* for Illumina (15 μM) to 1.5 μM
with a 10-fold dilution (1:9) with 10 mM Tris-HCl and 10 mM NaCl for immediate use.
(red) NEBNext Adaptor* for Illumina (15 μM) to 1.5 μM
with a 10-fold dilution (1:9) with 10 mM Tris-HCl and 10 mM NaCl for immediate use.
3.5.1.
Add the following components directly to the End Prep Reaction (Caution: Do not pre-mix the components to prevent adaptor-dimer formation):
End Prep Reaction 65 μl
(red) Blunt/TA Ligase Master Mix 15 μl
Diluted NEBNext Adaptor* 1 μl
Nuclease-free Water 2.5 μl
-----------------------------------------------------------------
Total volume 83.5 μl
*The adaptor is provided in NEBNext Singleplex (NEB #E7350) or NEBNext Multiplex (NEB #E7335, #E7500, #E7710, #E7730, #E6609 or #E7600) Oligos for Illumina.
3.5.2. Mix by pipetting followed by a quick spin to collect all liquid from the sides of the tube.
3.5.3. Incubate 15 minutes at 20°C in a thermal cycler.
A precipitate can form upon thawing of the NEBNext Hot Start HiFi PCR Master Mix. To ensure optimal performance, place the master mix at room temperature while purifying the ligation reaction. Once thawed, gently mix by inverting the tube several times.
3.6 Purify the Ligation Reaction Using AMPure XP Beads
Note: If you are selecting for larger size fragments (> 200 nt) follow the size selection recommendations in Appendix A, Chapter 4.
3.6.1. Add nuclease-free water to the ligation reaction to bring the reaction volume to 100 μl. It is important to ensure the final volume is 100 μl prior to adding AMPure XP Beads.
Note: X refers to the original sample volume of 100 μl from the above step.
3.6.2. Add 100 μl (1.0X) resuspended AMPure XP Beads and mix well on a vortex mixer or by pipetting up and down at least 10 times.
3.6.3. Incubate for 5 minutes at room temperature.
3.6.4. Quickly spin the tube in a microcentrifuge and place the tube on an appropriate magnetic rack to separate beads from the supernatant. After the solution is clear (about 5 minutes), discard the supernatant that contain unwanted fragments (Caution: do not discard the beads).
3.6.5. Add 200 μl of freshly prepared 80% ethanol to the tube while in the magnetic rack. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
3.6.6. Repeat Step 3.6.5 once for a total of 2 washing steps.
3.6.7. Briefly spin the tube, and put the tube back in the magnetic rack.
3.6.8. Completely remove the residual ethanol, and air dry beads for 5 minutes while the tube is on the magnetic rack with the lid open.
Caution: Do not over dry the beads. This may result in lower recovery of DNA target.
3.6.9. Remove the tube from the magnet. Elute DNA target from the beads with 52 μl 0.1X TE or 10 mM Tris-HCl. Mix well on a vortex mixer or by pipetting up and down, incubate for 2 minutes at room temperature. Put the tube in the magnetic rack until the solution is clear.
3.6.10. Transfer the 50 μl supernatant to a clean PCR tube. Discard beads.
3.6.11. To the 50 μl supernatant, add 50 μl (1.0X) of the resuspended AMPure XP Beads and mix well on a vortex mixer or by pipetting up and down at least 10 times.
3.6.12. Incubate for 5 minutes at room temperature.
3.6.13. Quickly spin the tube in a microcentrifuge and place the tube on an appropriate magnetic rack to separate beads from the supernatant. After the solution is clear (about 5 minutes), discard the supernatant that contains unwanted fragments (Caution: do not discard the beads).
3.6.14. Add 200 μl of freshly prepared 80% ethanol to the tube while in the magnetic rack. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
3.6.15. Repeat Step 3.6.14 once for a total of 2 washing steps.
3.6.16. Briefly spin the tube, and put the tube back in the magnetic rack.
3.6.17. Completely remove the residual ethanol, and air dry beads for 5 minutes while the tube is on the magnetic rack with the lid open.
Caution: Do not over dry the beads. This may result in lower recovery of DNA target.
3.6.18. Remove the tube from the magnet. Elute DNA target from the beads with 19 μl 0.1X TE or 10 mM Tris-HCl. Mix well on a vortex mixer or by pipetting up and down, incubate for 2 minutes at room temperature. Put the tube in the magnetic rack until the solution is clear.
3.6.19. Without disturbing the bead pellet, transfer 17 μl of the supernatant to a clean PCR tube and proceed to PCR enrichment.
3.7 PCR Enrichment of Adaptor Ligated DNA
Follow Section 3.7A if you are using the following oligos (10 μM primer):
NEBNext Singleplex Oligos for Illumina (NEB #E7350)
NEBNext Multiplex Oligos for Illumina (Set 1, NEB #E7335)
NEBNext Multiplex Oligos for Illumina (Set 2, NEB #E7500)
NEBNext Multiplex Oligos for Illumina (Set 3, NEB #E7710)
NEBNext Multiplex Oligos for Illumina (Set 4, NEB #E7730)
NEBNext Multiplex Oligos for Illumina (Dual Index Primers, NEB #E7600)
Follow Section 3.7B if you are using NEBNext Multiplex Oligos for Illumina (96 Index Primers, NEB #E6609)
3.7A PCR Library Enrichment
3.7A.1. To the cDNA (17 μl) from Step 3.6.19 add the following components and mix by gentle pipetting:
 (blue) NEBNext USER Enzyme 3 μl
(blue) NEBNext USER Enzyme 3 μl
(blue) NEBNext Q5 Hot Start HiFi PCR Master Mix 25 μl
(blue) Index (X) Primer/i7 Primer*,** 2.5 μl
(blue) Universal PCR Primer/i5 Primer*, *** 2.5 μl
-----------------------------------------------------------------------------------------------------
Total volume 50 μl
* The primers are provided in NEBNext Singleplex (NEB #E7350) or Multiplex (NEB #E7335, #E7500, #E7710, #E7730, #E7600) Oligos for Illumina. For use with Dual Index Primers (NEB #E7600), look at the NEB #E7600 manual for valid barcode combinations and tips for setting up PCR reactions.
** For use with NEBNext Multiplex Oligos (NEB #E7335, #E7500, #E7710, #E7730) use only one Index Primer per PCR reaction. For use with Dual Index Primers (NEB #E7600) use only one i7 Primer per reaction.
*** For use with Dual Index Primers (NEB #E7600) use only one i5 Primer per reaction.
3.7A.2. PCR Cycling Conditions
| CYCLE STEP | TEMPERATURE | TIME | CYCLES |
| USER Digestion Initial Denaturation |
37°C 98°C |
15 minutes 30 seconds |
1 1 |
| Denaturation Annealing/Extension |
98°C 65°C |
10 seconds 75 seconds |
12–15*, ** |
| Final Extension | 65°C | 5 minutes | 1 |
| Hold | 4°C | ∞ |
* The number of PCR cycles should be adjusted based on RNA input. If 10 ng enriched RNA is the starting input, it is recommended to perform 15 cycles of PCR. However, optimization of PCR cycle number may be required to avoid over-amplification.
** It is important to limit the number of PCR cycles to avoid overamplification. If overamplification occurs, larger molecular weight products (> 500 bp) will appear on the Bioanalyzer trace.
3.7A.3. Proceed to Section 3.8 (Purify the PCR Reaction using Agencourt AMPure XP Beads).
3.7B PCR Library Enrichment
3.7B.1. To the cDNA (17 μl) from Step 3.6.19 add the following components and mix by gentle pipetting.
(blue) NEBNext User Enzyme 3 μl
(blue) NEBNext Q5 Hot Start HiFi PCR Master Mix 25 μl
(blue) Index/ Universal Primer Mix* 5 μl
-----------------------------------------------------------------------------------------------------
Total volume 50 μl
* The primers are provided in NEBNext Multiplex Oligos for Illumina, NEB #E6609. Please refer to the NEB #E6609 manual for valid barcode combinations and tips for setting up PCR reactions.
3.7B.2. PCR Cycling Conditions
| CYCLE STEP | TEMPERATURE | TIME | CYCLES |
| USER Digestion | 37°C | 15 minutes | 1 |
| Initial Denaturation | 98°C | 30 seconds | 1 |
| Denaturation Annealing/Extension |
98°C 65°C |
10 seconds 75 seconds |
12–15*, ** |
| Final Extension | 65°C | 5 minutes | 1 |
| Hold | 4°C | ∞ |
* The number of PCR cycles should be adjusted based on RNA input. If 10 ng enriched RNA is the starting input, it is recommended to perform 15 cycles of PCR. However, optimization of PCR cycle number may be required to avoid over-amplification.
** It is important to limit the number of PCR cycles to avoid overamplification. If overamplification occurs, larger molecular weight products (> 500 bp) will appear on the Bioanalyzer trace.
3.7B.3. Proceed to Section 3.8 (Purify the PCR Reaction using Agencourt AMPure XP Beads).
3.8 Purify the PCR Reaction using Agencourt AMPure XP Beads
3.8.1. Vortex Agencourt AMPure XP Beads to resuspend.
3.8.2. Add 45 μl (0.9X) of resuspended Agencourt AMPure XP Beads to the PCR reaction (~ 50 μl). Mix well on a vortex mixer or by pipetting up and down at least 10 times.
3.8.3. Incubate for 5 minutes at room temperature.
3.8.4. Quickly spin the tube in a microcentrifuge and place the tube on an appropriate magnetic rack to separate beads from the supernatant. After the solution is clear (about 5 minutes), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain DNA targets.
3.8.5. Add 200 μl of freshly prepared 80% ethanol to the tube while in the magnetic rack. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
3.8.6. Repeat Step 3.8.5 once for a total of 2 washing steps.
3.8.7. Air dry the beads for 5 minutes while the tube is on the magnetic rack with the lid open.
Caution: Do not overdry the beads. This may result in lower recovery of DNA target.
3.8.8. Remove the tube from the rack. Elute the DNA target from the beads into 23 μl 0.1X TE. Mix well on a vortex mixer or by pipetting up and down, quickly spin the tube in a microcentrifuge and incubate for 2 minutes at room temperature. Place it in the magnetic rack until the solution is clear.
3.8.9. Transfer 20 μl of the supernatant to a clean PCR tube, and store at –20°C.
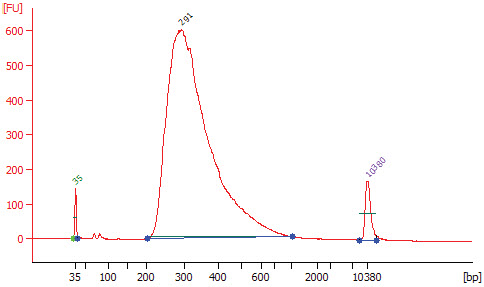
3.9 Assess library quality on a Bioanalyzer (Agilent High Sensitivity Chip).
3.9.1. Dilute 2–3 μl of the library in 10 mM Tris or 0.1X TE.
3.9.2. Run 1 μl in a DNA High Sensitivity Chip.
3.9.3. Check that the electropherogram shows a narrow distribution with a peak size approximately 300 bp.
Note: If a peak at ~ 80 bp (primers) or 128 bp (adaptor-dimer) is shown in the Bioanalyzer traces, bring up the sample volume (Step 3.8.9) to 50 μl exactly with nuclease-free water and repeat the AMPure XP Bead clean up step (Section 3.8).